Role of the noncoding genome in de novo gene birth and protein evolution
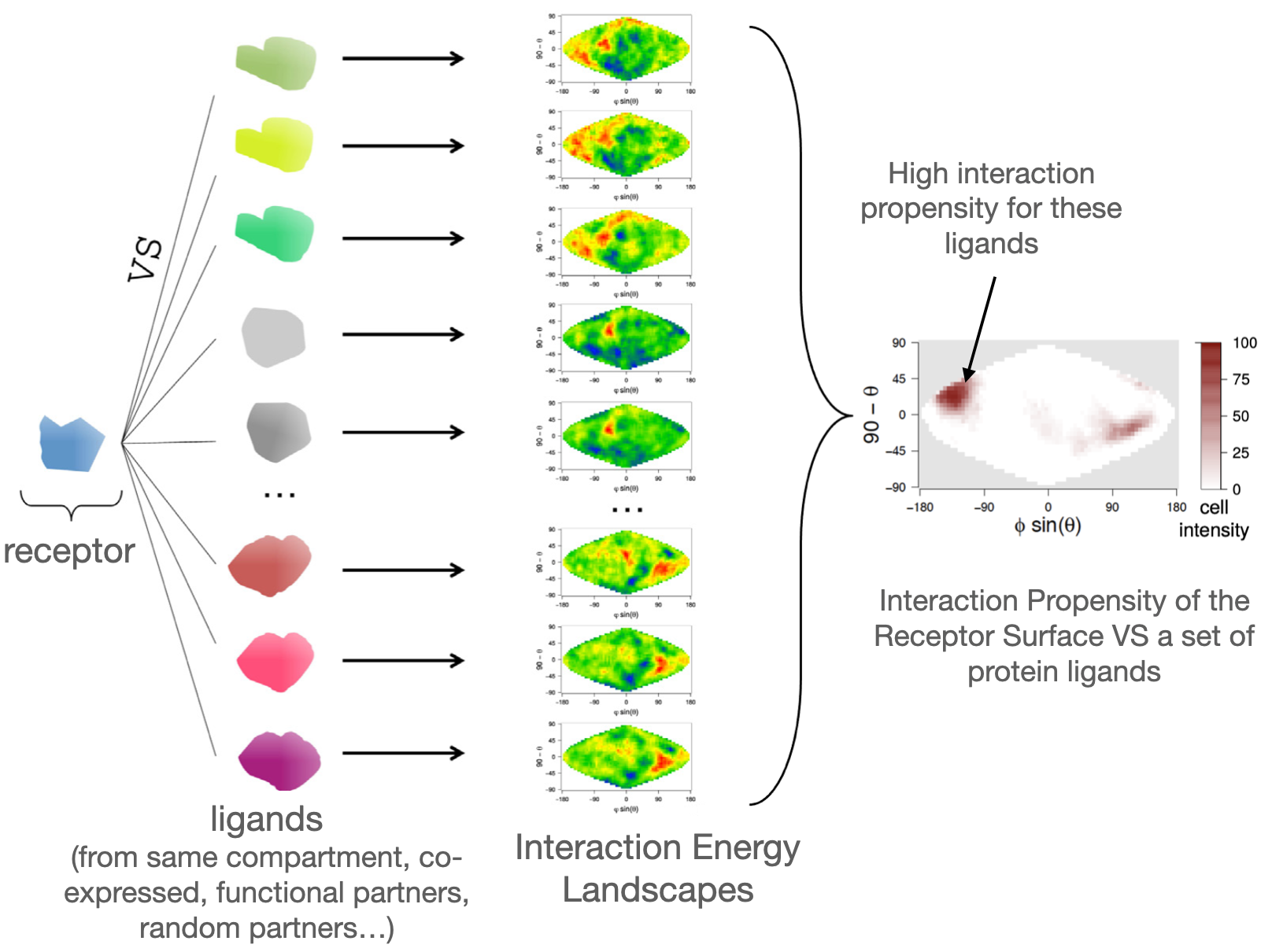
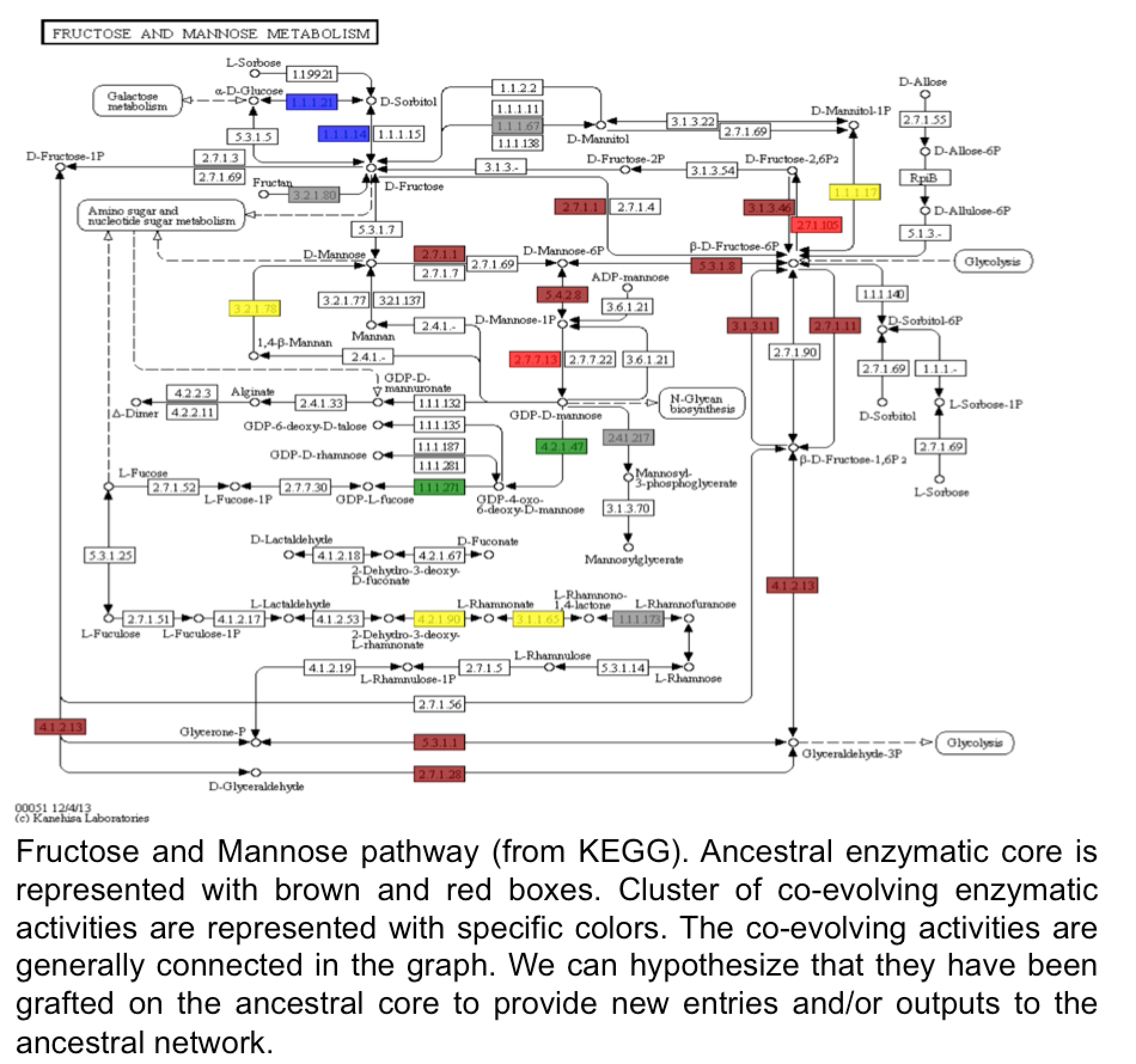
Today it is well known that the noncoding genome provides the organisms with the raw material for novel RNAs and protein products, thus playing an important role in the emergence of genetic novelty. The wide use of transcriptomics has revealed a high pervasive transcription of presumed noncoding regions, and a fraction of the resulting RNAs have been shown to be translated by ribosome profiling experiments. If an important fraction of them is expected to be degraded right-away, some of them may be neutral or deleterious and, therefore, will be short-lived in evolutionary history, while a small fraction of them, if they provide an advantage to the organism, will be fixed and established as novel genes. These results attribute a central role to the noncoding genome in the emergence of new genes and the development of pathologies. However, the mechanisms governing these processes remain unclear, though they are essential to understand (i) the evolutionary forces governing the emergence of these new genes and the evolution of genomes and (ii) their role in some cancers, or other human pathologies. In this project, we aim at investigating with integrative approaches the potential of the noncoding genome to give rise to novel peptides or novel protein bricks in order to better understand its role in the evolution of organisms and the development of human diseases.